
FDA makes clearance easier for cardiac device implant simulation software
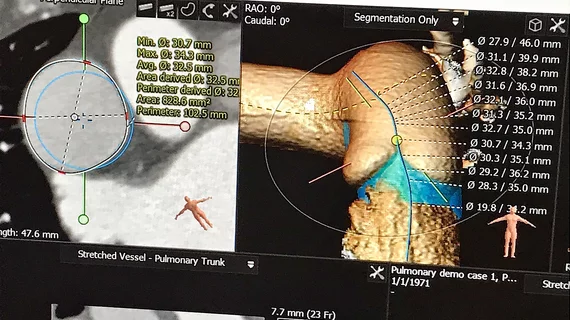
The U.S. Food and Drug Administration (FDA) is reclassifying interventional cardiovascular implant simulation software into the class II (special controls) regulatory category, which will make it easier for the technology to pass regulatory approval. The technology has grown rapidly over the last few years to better assess the fit and possible complications from device implants as part of the regular procedure pre-planning.
"We are taking this action because we have determined that classifying the device into class II (special controls) will provide a reasonable assurance of safety and effectiveness of the device. We believe this action will also enhance patients' access to beneficial innovative devices," the FDA said in its Federal Register report.
As with any new technologies that come before the FDA, all automatically fall under Class III, which requires premarket approval (PMA). The FDA can take an action to classify or reclassify the device into less strenuous regulatory approval tracks based on reasonable safety review, which is what the FDA has done with the new simulated implant technology.
The FDA order became effective as a final order Dec. 28, 2022. Read the new FDA rule.
Virtual cardiac device implant software has seen rapid adoption
This technology area has seen its greatest amount of use in structural heart procedures, where devices can be placed in a virtual 3D simulation of the patient's anatomy using a computed tomography (CT) scan. This is often done in left atrial appendage occlusion (LAAO) and transcatheter valve procedures to check device sizing and fit in various patient anatomies to prevent embolization and ensure complete seal of the LAA. These simulations can also help assess access routes for these larger delivery catheters by identifying sharp bends and potential issues with blood flow obstruction.
In the case of transcatheter mitral valve replacements (TMVR), the simulation software can show if there will be left ventricular outflow tract (LVOT) obstruction. This can help avoid a potentially fatal obstruction, or determine if other procedures can be used to modify the anatomy prior to the implant.
A recent area of device implant simulation development has been virtual coronary stent placement. One company that has FDA clearance for this technology is Heartflow, which uses computational fluid dynamics to assess if opening a vessel segment with a stent will improve hemodynamic flow. This also can be used to test locations of multiple stents in long-diffuse coronary disease and may aid procedural planning.
All of the above cardiovascular device implant simulation technologies are discussed on a regular basis in cardiology conference sessions, including "how-to" courses on its use at some meetings.
What is required for FDA clearance of cardiac device implant simulation software?
Under the Class II (special controls) guidelines, the following are required:
1. Software verification, validation and hazard analysis, with identification of appropriate mitigations, must be performed. This includes a full verification and validation of the software according to the predefined software specifications.
2. Computational modeling verification and validation activities must be performed to establish the predictive capability of the device for its indications for use.
3. Performance validation testing must be provided to demonstrate the accuracy and clinical relevance of the modeling methods for the intended implantation simulations, including the following:
• Computational modeling results must be compared to clinical data supporting the indications for use to demonstrate accuracy and clinical meaningfulness of the simulations.
• Agreement between computational modeling results and clinical data must be assessed and demonstrated across the full intended operating range (such as the full range of patient population, implant device sizes and patient anatomic morphologies). Any selection criteria or limitations of the samples must be described and justified;
• Endpoints such as performance goals and sample sizes established must be justified as to how they were determined and why they are clinically meaningful.
• Validation must be performed and controls implemented to characterize and ensure consistency.

Dave Fornell has covered healthcare for more than 17 years, with a focus in cardiology and radiology. Fornell is a 5-time winner of a Jesse H. Neal Award, the most prestigious editorial honors in the field of specialized journalism. The wins included best technical content, best use of social media and best COVID-19 coverage. Fornell was also a three-time Neal finalist for best range of work by a single author. He produces more than 100 editorial videos each year, most of them interviews with key opinion leaders in medicine. He also writes technical articles, covers key trends, conducts video hospital site visits, and is very involved with social media. E-mail: [email protected]