
FDA announces new Class I recall of drainage systems after 26 injuries

The U.S. Food and Drug Administration (FDA) has announced that Medtronic is recalling more than 45,000 cerebrospinal fluid (CSF) drainage systems due to a risk of the catheter disconnecting during aortic aneurysm surgeries.
This is a Class I recall, which means using these devices “may cause serious injuries or death.”
Medtronic’s Duet External Drainage and Monitoring System (EDMS) is used to drain or sample CSF during open descending thoracic aortic aneurysm (open TAA) or open descending thoraco-abdominal aortic aneurysm (open TAAA) surgeries. They may also be used following surgery if patients develop symptoms such as paraplegia. The device works by using gravity to carry CSF from an external lumbar catheter through a patient tube and a drip chamber before finally filling a removable bag.
Medtronic is recalling the Duet EDMS due to a risk of the catheter disconnecting from the rest of the device.
“If a tubing disconnection occurs, potential harm to patients may include infections, cerebrospinal fluid leakage, over drainage of cerebrospinal fluid, and abnormality of the ventricles,” the FDA warns. “Uncontrolled over drainage of cerebral spinal fluid could lead to neurological injury or death if the disconnection is undetected.”
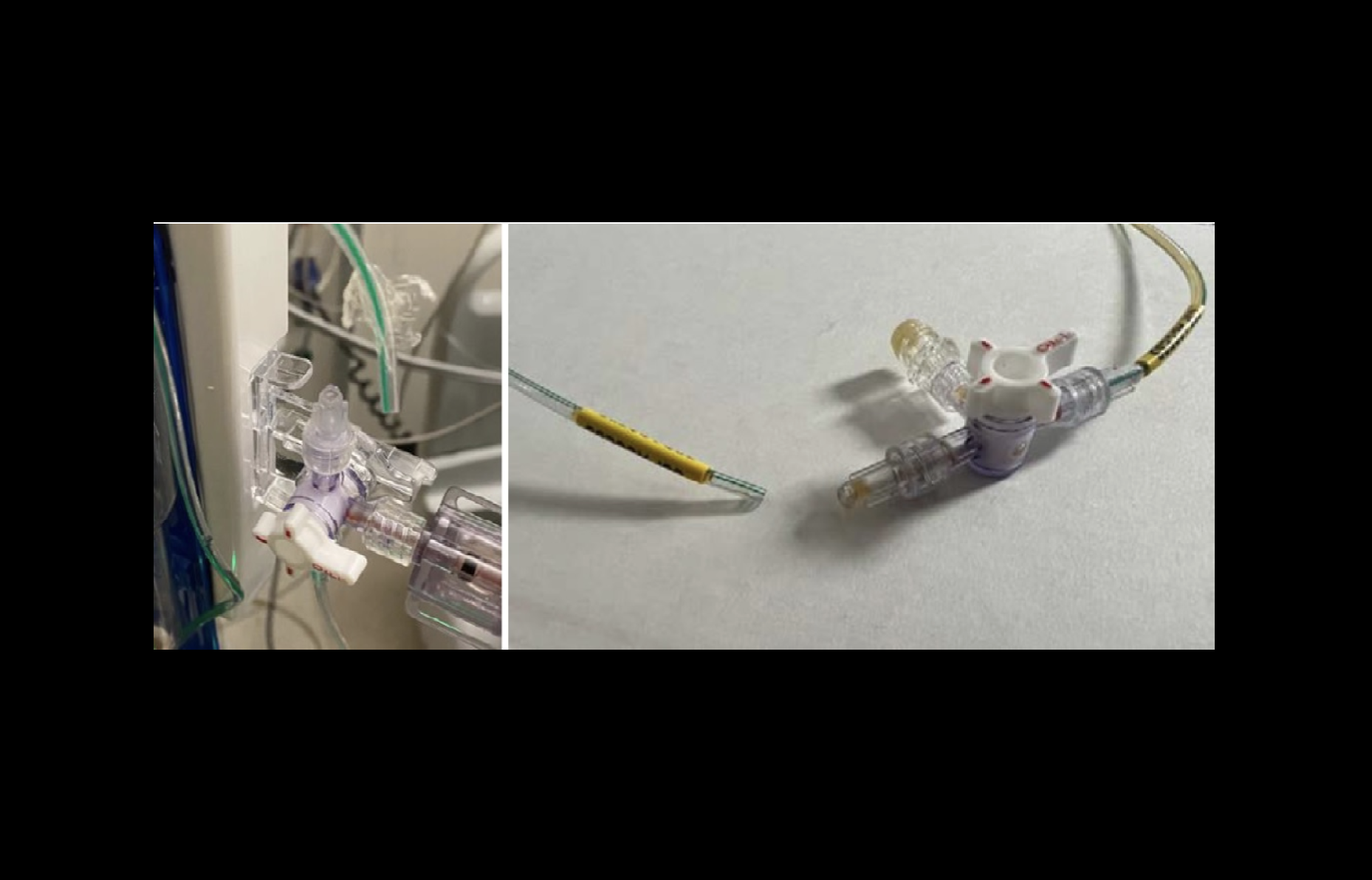
Medtronic is recalling the Duet EDMS due to a risk of the catheter disconnecting from the rest of the device. Image courtesy of Medtronic.
A total of 26 injuries have been reported so far due to this issue. There have been no patient deaths.
This Class I recall includes a total of 45,176 devices that were first distributed to customers beginning in May 2021. Model numbers of the impacted drainage systems are 46913, 46914, 46915, 46916, and 46917.
If a patient is being actively treated with a Duet EDMS and a leak or disconnection is identified, Medtronic recommends the device be replaced with a new one. If no leak or disconnection is detected, treatment can continue as intended.
Unused products included in this recall, on the other hand, can be returned to Medtronic.
Click here to read the full advisory on the FDA’s website.

Michael has more than 19 years of experience as a professional writer and editor. He has written at length about cardiology, radiology, artificial intelligence and other key healthcare topics.