
FDA finalizes Class I recall for Medtronic catheters
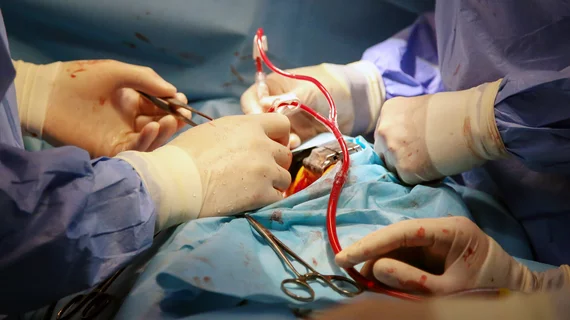
The U.S. Food and Drug Administration (FDA) has confirmed a new recall for certain cannulae used during cardiopulmonary bypass surgery procedures. The recall includes dozens of lots of Medtronic’s DLP Left Heart Vent Catheters with malleable bodies and vented connectors.
Medtronic is asking customers to quarantine all unused devices in their inventory and return them to the company.
The recall was put in place due to ongoing safety concerns. The FDA first alerted the public about the issue in August after receiving multiple reports of the catheters “resisting shape retention when being bent.” While the devices were designed to be easy to bend, it can cause significant delays in care if they fail to hold their shape.
“If the issue is not identified prior to use and the clinician uses the cannula, it may lead to abrasion and perforation (cuts),” the FDA warned at the time. “Perforation of critical heart tissue may potentially lead to death if it is complicated, unnoticed or untreated.”
When the FDA issued that early warning, it was still reviewing the situation. Now, the agency has officially determined this is a Class I recall, which means using the device increases the risk of serious injuries or death.
This issue has been linked to three serious injuries and no patient deaths.
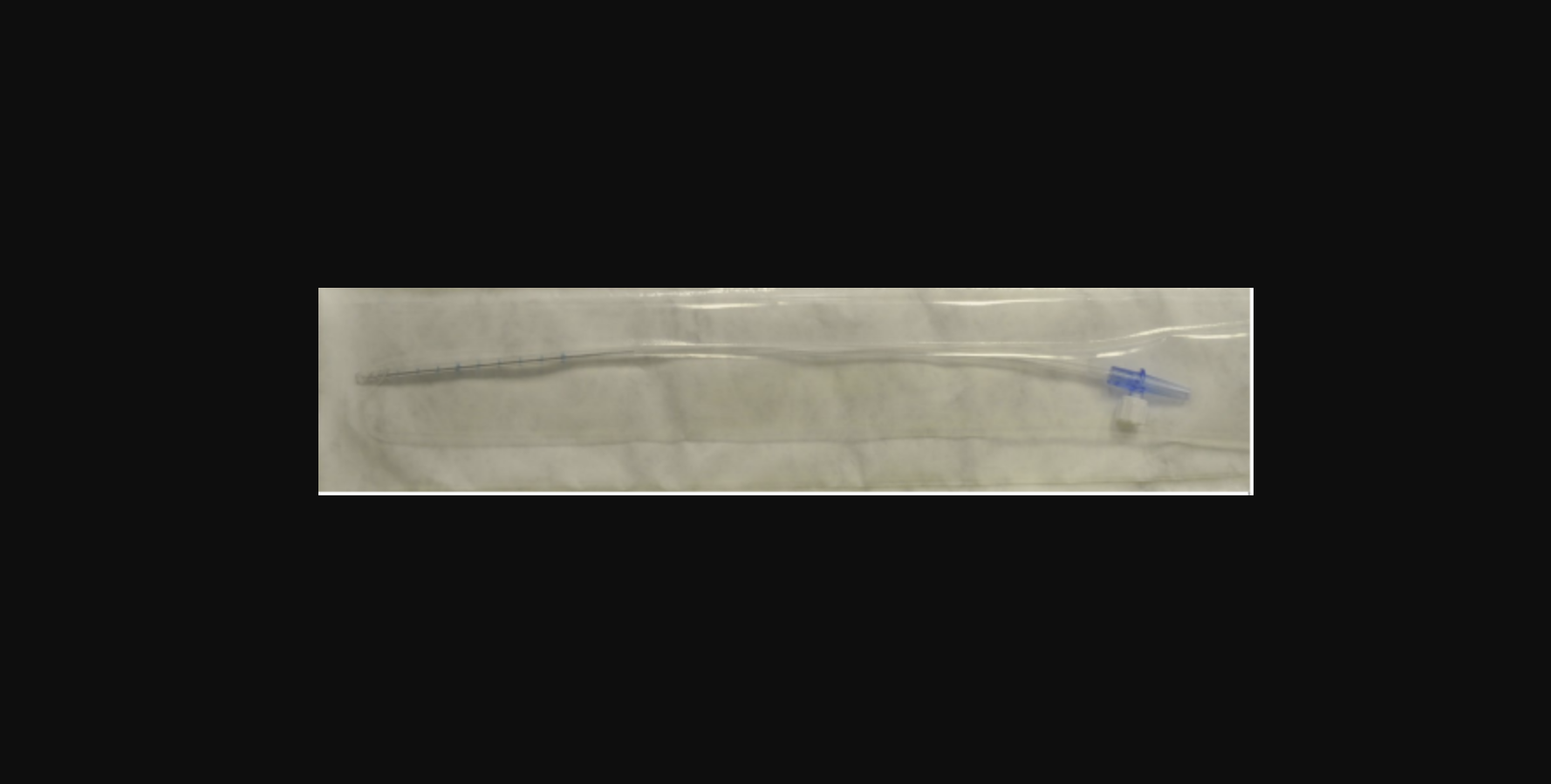
One of Medtronic's DLP Left Heart Vent Catheters. Image courtesy of the FDA.
FDA’s warning about a potential device shortage
In its advisory about this recall, the FDA noted that Medtronic could have a limited supply of these devices until more can be made available.
“If the product is unavailable, you may work with your sales representative to explore potential replacement options that Medtronic can offer,” according to the agency. “Alternatively, Medtronic will issue a credit note if a suitable replacement is not available.”

Michael has more than 19 years of experience as a professional writer and editor. He has written at length about cardiology, radiology, artificial intelligence and other key healthcare topics.