
Medline recalls millions of devices due to safety risk—FDA threatens ‘regulatory action’ in warning letter

Medline, an Illinois-based medical supplier that went public in December 2025, has recalled millions of NAMIC angiographic control syringes due to a significant safety concern.
These syringes, often included in procedure kits, are used in cardiac cath labs or interventional radiology suites for the intra-arterial or intravenous administration of radiographic contrast media. Now, the devices—and several kits that feature them—are officially part of a Class I recall due to a risk of a “a loose connection and/or full disconnection between the syringe and manifold.”
Customers have been told to stop using these syringes immediately and destroy any that happen to be on hand. The only exception would be cases when angiography is “urgently required” and no alternative devices are available.
According to the FDA, this recall includes more than 4 million syringes and more than 1 million procedure kits that contain the syringes. Exact lot details are available from the FDA.
FDA’s letter to Medline highlights multiple violations
Medline received a warning letter from the U.S. Food and Drug Administration (FDA) detailing multiple violations related to these same devices.
According to the letter, dated March 25 and later published online, an inspection of a Medline facility in New York revealed that the devices had been “adulterated” in a way that was not up to current quality standards.
The FDA had already shared its concerns with Medline in a previous communication. However, the agency was not satisfied with Medline’s initial response; that prompted this warning letter to be sent and then shared with the public.
FDA not satisfied with Medline’s response to safety concerns
The FDA’s letter cited several violations. For instance, the agency said Medline failed to properly respond to “an increase in complaints” about these syringes and manifolds that were first reported in June 2023. According to the FDA, Medline’s own assessments of the risks associated with these devices were inconsistent and the company did not do enough to ensure the problem—which is believed to be due to excess silicone—would not reoccur.
The FDA specifically called out two complaints it observed about these devices, including one involving “the injection of air into a patient” and another involving “biohazard exposure of a clinician.” Four injuries associated with the syringes have been reported so far.
The FDA also wrote that it is still waiting on more information about Medline’s efforts to investigate these issues.
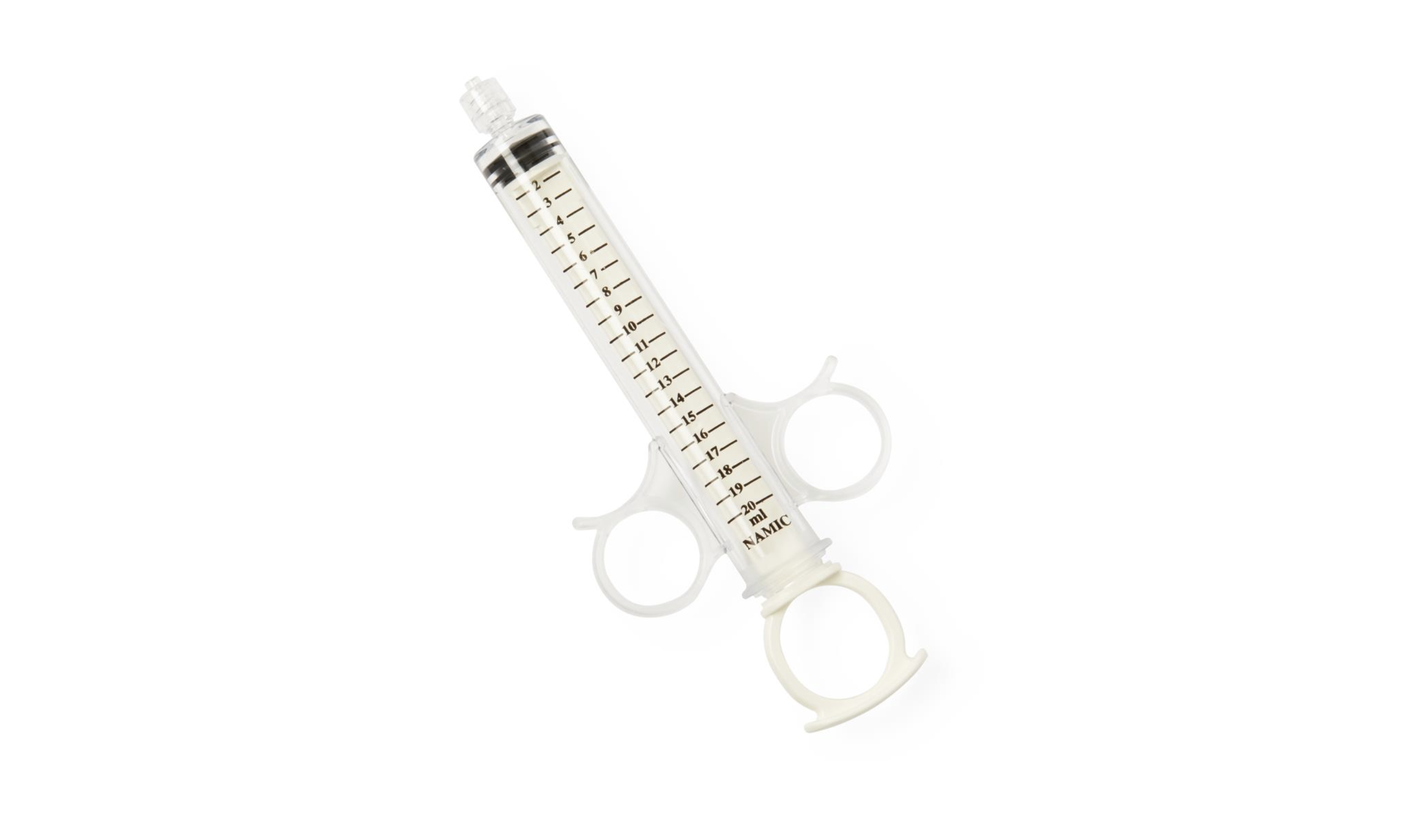
Medline's Namic Angiographic 12 mL Control Syringe with Rotator. Image courtesy of Medline.
FDA shares additional concerns
Another key issue brought up in the warning letter is Medline’s “failure to establish and maintain schedules for the adjustment, cleaning and other maintenance of equipment to ensure that manufacturing specifications are met.” As one example of this issue, the FDA noted that there were more than 100 complaints related to “foreign matter or hair in the package or device” from December 2023 to December 2025.
Again, the FDA brought this concern up with Medline previously, but it says the company’s response was “not adequate.” While Medline did say it has finalized a plan to maintain better environments going forward, the FDA claims the company did not properly explain how it would ensure devices that have already gone through those environments will not create any safety issues.
Another issue mentioned in the FDA’s warning letter involves the way Medline verified the design of these devices. According to the agency, Medline’s design verification failed to demonstrate that the polycarbonate female luer connector components of its fluid management product lines met the latest standards after undergoing a necessary update.
FDA urges Medline to act quickly
The FDA closed its letter by asking Medline to “take prompt action” and address these violations. The agency emphasized that other federal agencies may take Medline’s actions into account when considering federal contracts.
“Failure to adequately address this matter may result in regulatory action being initiated by the FDA without further notice,” according to the letter. “These actions include, but are not limited to, seizure, injunction and civil money penalties.”

Michael has more than 19 years of experience as a professional writer and editor. He has written at length about cardiology, radiology, artificial intelligence and other key healthcare topics.