
FDA warns against use of troubled heart devices, citing ‘continued concerns’ after multiple recalls

The U.S. Food and Drug Administration (FDA) is urging hospitals and other healthcare facilities to stop using certain Getinge products and transition to medical devices developed by other companies. According to the agency, the company has failed to address significant safety concerns that resulted in multiple Class I recalls.
The devices in question include the Cardiosave Hybrid and Rescue intra-aortic balloon pumps (IABPs) manufactured by Datascope, a subsidiary of Getinge, and the Cardiohelp extracorporeal membrane oxygenation (ECMO) systems.
“The FDA recommends that health care facilities transition away from use of these devices and seek alternatives, if possible,” according to the agency’s warning.
The Cardiosave Rescue Intra-Aortic Balloon Pump.
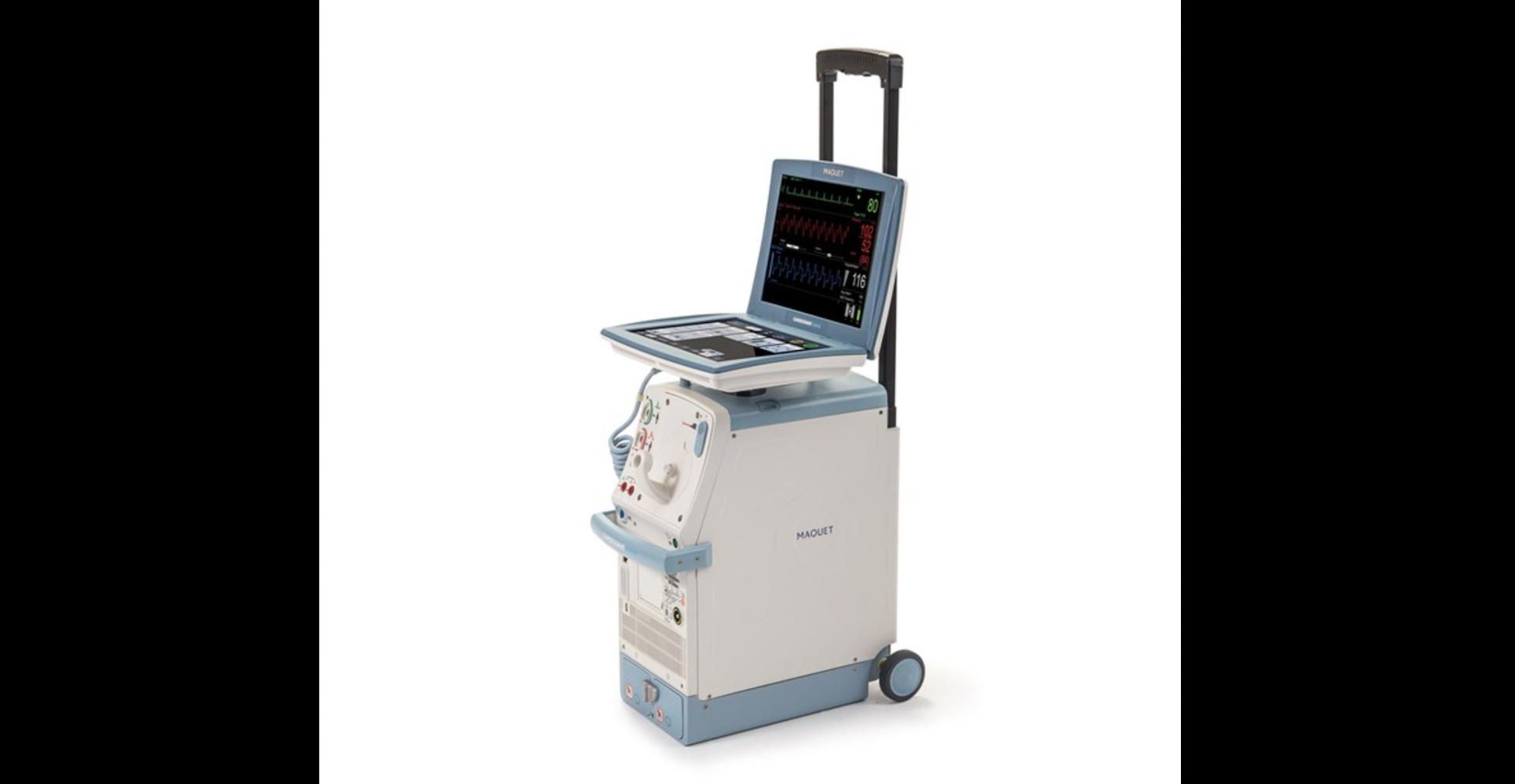
Cardiosave IABPs
The Cardiosave Hybrid and Rescue IABPs, designed to provide temporary support to a patient’s left ventricle, have been involved in a total of 12 voluntary recalls from January 2023 to April 2024. Eight of those were Class I recalls, which means the FDA believed using the devices could have resulted in “serious injuries or death.”
The FDA said it received nearly 3,000 medical device reports related to the Cardiosave IABPs in the last 12 months. Fifteen of those reports were related to a serious injury or death, though the agency did emphasize it can be “difficult to confirm a direct cause and effect.”
The consistent issue with these devices has been that they unexpectedly stop working during use. When this happens, it creates a risk of interrupting patient care in a way that could potentially lead to an adverse event.
Some of the reasons the devices are shutting down include electrical failures in the power management board or solenoid board, failures in the printed circuit board assembly in the charging path, power failures due to the devices being placed incorrectly into their carts, failure alarms due to helium issues, sensitive “gas gain” and “gas loss” alarms going off and overheating.
In September 2023, the FDA compiled all of the relevant details up to that point into a single open letter for healthcare providers. Click here for more information.

The Cardiohelp extracorporeal membrane oxygenation (ECMO) system and its emergency drive (at top of device).
Cardiohelp ECMO systems
The Cardiohelp ECMO devices, designed to help pump blood out of a patient during cardiopulmonary bypass surgeries, have been involved in eight voluntary recalls from January 2023 to April 2024. One of those was a Class I recall.
The FDA said it received 246 medical device reports related to the Cardiohelp ECMO systems over the last 12 months. Thirty-three of those reports were related to a serious injury or death.
One of the biggest issues was the fact that the emergency drive’s handle was difficult to turn. In some cases, it became completely stuck.
Click here for additional context about the Class I recall announced last December.
How the FDA is responding
“The FDA continues to work with the Getinge to understand factors contributing to the device failures, as well as possible mitigation strategies,” according to the warning to healthcare providers. “The FDA is working with other manufacturers to assess their interest and ability to manufacture and distribute alternative devices in the U.S.”
The agency also emphasized that it will continue to keep the public informed about these concerns.

Michael has more than 19 years of experience as a professional writer and editor. He has written at length about cardiology, radiology, artificial intelligence and other key healthcare topics.