
FDA panel votes against approving first-of-its-kind heart failure device
A U.S. Food & Drug Administration (FDA) advisory panel has voted unanimously that the benefits associated with a new heart failure device from Johnson & Johnson MedTech do not outweigh the risks.
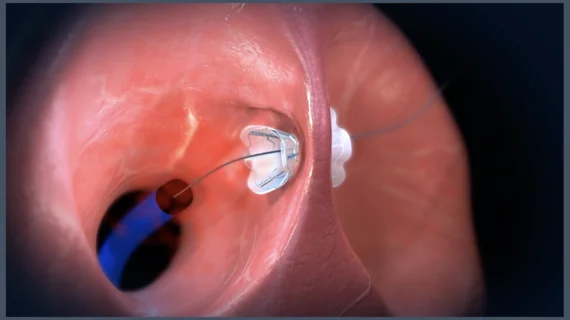
The device in question is the V-Wave Ventura Interatrial Shunt System, a permanent implant that previously gained the FDA’s breakthrough device designation for its potential to address an unmet need. The shunt was designed to treat heart failure patients by reducing left atrial pressure, and it is largely seen as the first implantable heart failure device of its kind.
The 15-person Circulatory System Devices Panel reviewed data from RELIEVE-HF, a randomized trial of 206 patients with heart failure with reduced ejection fraction (HFrEF) and 302 patients with heart failure with preserved ejection fraction (HFpEF). Patients were either treated with the V-Wave shunt or a placebo procedure. Patients and healthcare providers were blinded to the treatment strategy for the full duration of the two-year study.
Overall, RELIEVE-HF did meet its primary safety endpoint. There were no major adverse cardiovascular or neurological events after 30 days or 24 months. The study’s primary effectiveness endpoint, however, was neutral due to the fact that HFrEF patients experienced improved outcomes and HFpEF patients actually experienced worse outcomes.
This difference between HFrEF and HFpEF patients was unexpected and certainly hurts the device’s odds of being approved. In addition, RELIEVE-HF did not reveal significant improvements in quality of life after two years when tracking Kansas City Cardiomyopathy Questionnaire overall summary scores. The study’s authors pointed to these issues in their own analysis, which was published in Circulation back in September 2024.[1]
Reviewing the panel’s decisions
In addition to its ruling that the benefits of treatment did not outweigh the risks, the advisory panel voted unanimously that there was not “reasonable assurance” the device was truly effective. However, the group did rule that the device was safe for patients by a vote of 9 to 6.
“The sponsors ran a well-executed study, and the FDA and our panelists did a great job navigating this complicated statistical analysis,” panelist Amit Shanker, MD, an attending physician with the St. Lawrence Health System, told Cardiovascular Business. “From our perspective, the issue with this subgroup analysis was that it’s a limited number of people without achieving statistical significance to really confirm the effectiveness of the device. The signal is favorable, but we still need more data.”
The FDA typically follows the guidance of these advisory panels, but that is not always the case. It is still possible the V-Wave Ventura Interatrial Shunt System will ultimately secure FDA approval.
“If the FDA does approve it, it would be just for the HFrEF population,” Shanker said. “If they don’t approve it, I imagine they’ll be requesting a larger randomized trial to look at this a bit more closely and have a larger sample size.”
Johnson & Johnson’s history with this device
Johnson & Johnson acquired V-Wave, the company that first developed this technology, back in 2024. While the initial purchase price was $600 million, the final total could reach approximately $1.7 billion if certain regulatory and commercial milestones are met. Once the acquisition was finalized, V-Wave officially became a part of Johnson & Johnson MedTech.

Michael has more than 19 years of experience as a professional writer and editor. He has written at length about cardiology, radiology, artificial intelligence and other key healthcare topics.