
New recalls announced for troubled heart devices—FDA previously urged customers to seek alternatives

The U.S. Food and Drug Administration (FDA) has finalized new recalls for the Cardiosave Hybrid and Rescue intra-aortic balloon pumps (IABPs) manufactured by Datascope, a subsidiary of Getinge. This includes a total of two Class II recalls that cover more than 11,500 IABPs in total.
These recalls do not require the devices to be removed from facilities or returned to the manufacturer. Instead, the goal is to ensure all customers are aware of four specific issues and review the updated instructions for use (IFU).
The recalls focus on four separate field safety correction actions related to these devices:
- Preventive maintenance related to the non-volatile random access memory (NVRAM) battery. Any NVRAM batteries more than eight years old could potentially lose their charge. This issue has been linked to a total of 85 customer complaints, but no adverse outcomes. It is recommended to keep backup batteries on hand.
- Vibration and shock table specifications. The IFU associated with these devices included the wrong transportation standards. This has not been linked to any issues at this time.
- Helium O-ring preventive maintenance. Certain O-rings should be replaced after approximately 12 months. This issue, previously addressed in a 2023 letter to customers, is now a part of the updated IFU.
- Battery runtime specifications. These devices have a known history of battery issues—click here or here for examples—and previous recalls have been completed. However, Datascope/Getinge emphasized that the updated IFU now includes the latest details on all battery specifications.
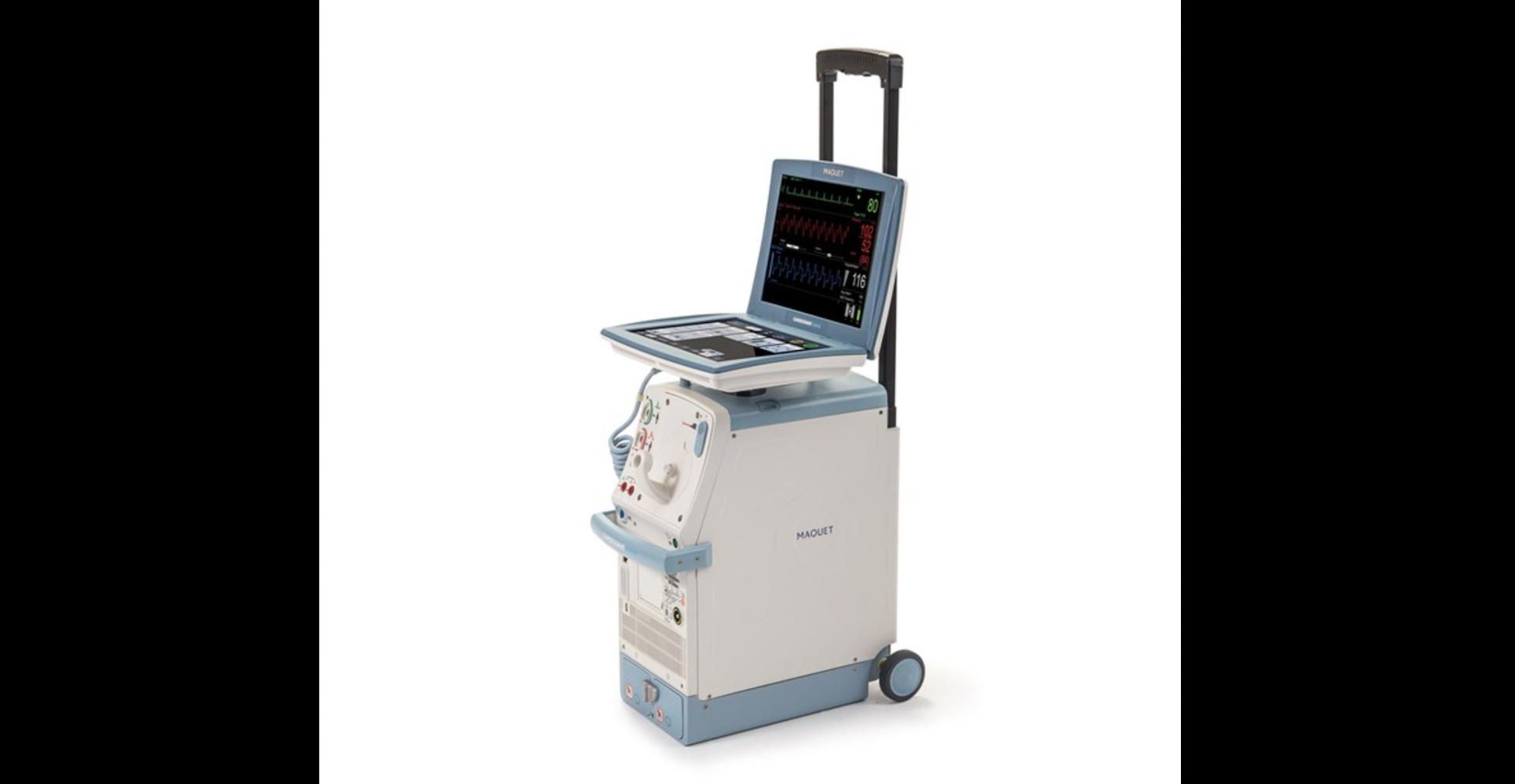
The Cardiosave Rescue Intra-Aortic Balloon Pump.
FDA’s 2024 warning about the Cardiosave Hybrid and Rescue intra-aortic balloon pumps
The Cardiosave Hybrid and Rescue IABPs, designed to provide temporary support to a patient’s left ventricle, were involved in a total of 12 voluntary recalls from January 2023 to April 2024. Eight of those were Class I recalls, which means the FDA believed using the devices could have resulted in “serious injuries or death.”
A common theme for these recalls was that the devices would unexpectedly stop working. Some of the reasons the devices were shutting down included electrical failures in the power management board or solenoid board, failures in the printed circuit board assembly in the charging path, power failures due to the devices being placed incorrectly into their carts, failure alarms due to helium issues, sensitive “gas gain” and “gas loss” alarms going off and overheating.
In May 2024, in the wake of those recalls, the FDA issued a warning that urged hospitals and other healthcare facilities to stop using the Cardiosave Hybrid and Rescue IABPs. The agency specifically recommended transitioning away from these devices and seeking alternative treatment options.
History of issues resulted in temporary suspension of CE mark approval in Europe
These safety concerns also impacted Datascope/Getinge in Europe. The company had its CE mark approval for the Cardiosave IABP technology suspended for more than a year before it was reinstated in August 2025.
“We are aware of the challenges the CE-mark suspension has caused for healthcare professionals and patients,” Elin Frostehav, president of acute care therapies at Getinge, said at the time. “Our teams have worked persistently to adhere to the required corrective actions in close dialogue with our Notified Body, TÜV SÜD.”

Michael has more than 19 years of experience as a professional writer and editor. He has written at length about cardiology, radiology, artificial intelligence and other key healthcare topics.